اعثر على اجابات على أسئلتك الشائعة المتعلقة بمرض نقص ألفا مانوسيداز. تم ترتيب الأسئلة تبعًا للموضوع من أجل تيسير عملية البحث. مرر المؤشر فوق كل سؤال للعثور على الإجابة التي تحتاجها.
مرض نقص ألفا مانوسيداز هو مرض وراثي نادر يمكن أن يصيب الأطفال والبالغين بتشوهات الهيكل العظمي وخشونة ملامح الوجه وفقدان السمع والتأخر العقلي ومشاكل في جهاز المناعة واضطرابات نفسية ومشاكل سلوكية.1 عندما لا يعمل الجين الذي يصدر التعليمات لتصنيع إنزيم الألفا مانوسيداز بشكل سليم، لا يتم إنتاج الإنزيم بشكل صحيح، مما يتسبب في الإصابة بمرض نقص ألفا مانوسيداز.2 يعمل هذا الإنزيم في الجسيمات الحَالّة، وهي عبارة عن حجرات تهضم المواد وتعيد تدويرها داخل الخلية. يساعد الإنزيم في تكسير سلاسل طويلة من جزيئات السكر (السكريات قليلة التعدد) داخل الجسيمات الحالة. تُستخدم السكريات قليلة التعدد في بناء العظام والغضاريف والجلد والأوتار والعديد من الأنسجة الأخرى في الجسم.3 4 عند الأشخاص المصابين بمرض نقص ألفا مانوسيداز، تظل السكريات المحللة جزئيًا مخزنة في الجسم وتتراكم بمرور الوقت. مما يتسبب في زيادة تضرر الخلايا.5قد يظهر على الأطفال القليل من علامات المرض ولكن مع تزايد الخلايا المتضررة بسبب تراكم السكريات قليلة التعدد، تبدأ الأعراض في الظهور.6
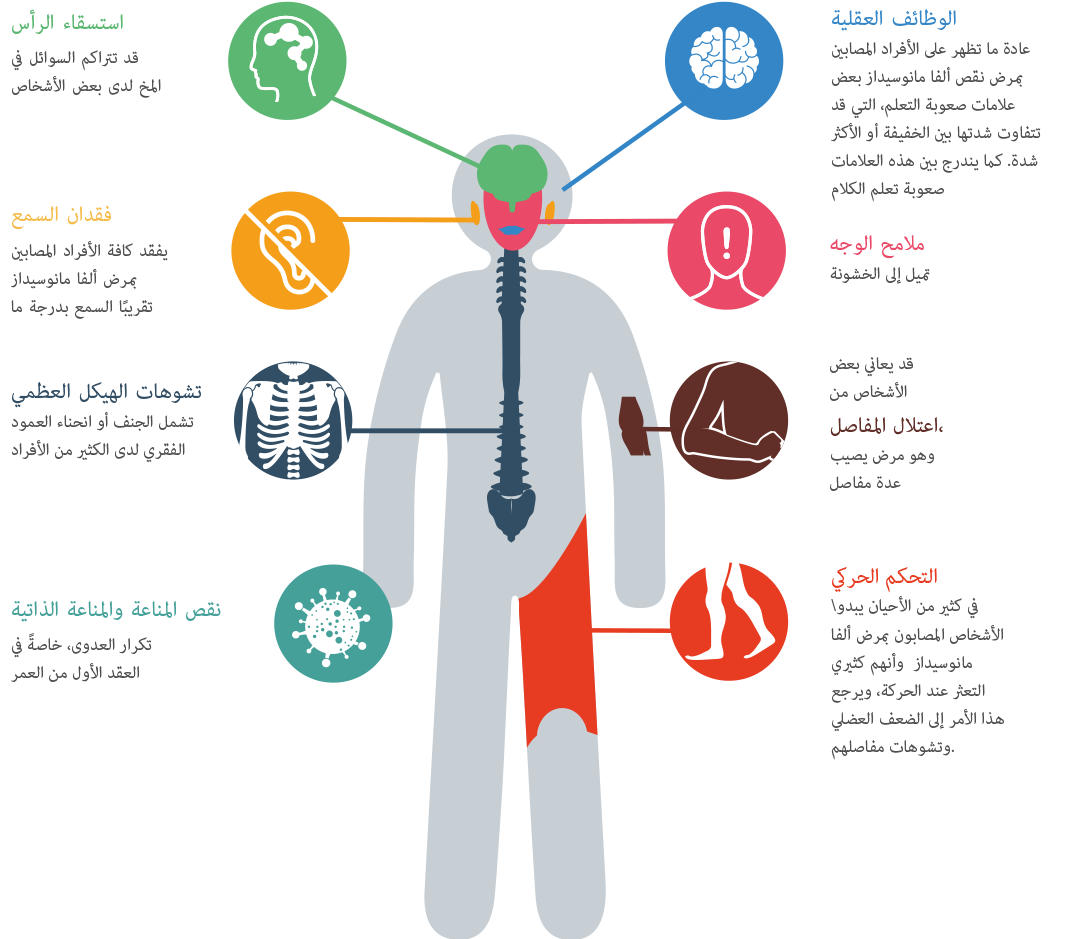
يُمكن أن تكون أعراض مرض نقص ألفا مانوسيداز شديدة التغير، كغيره من الاضطرابات الأخرى ذات الصلة .7 فهو مرض تدريجي يظهر بعدة أشكال على مدار فترة من الزمن.8 نظرًا لأن له مجموعة واسعة من الأعراض، ولأن كل فرد سوف يعاني من نمطه الفريد من الأعراض، يمكن أن يستغرق التشخيص وقتًا طويلاً.9 فخلال العقد الأول من العمر، قد يعاني الطفل المصاب بهذه الحالة من التهابات متكررة ومشاكل السمع وملامح الوجه المميزة وتأخر النمو.10 11 قد يلاحظ الآباء أن طفلهم قد يعاني من بعض الضعف العضلي.12 قد تظهر بعض الملامح غير الطبيعية مثل اعوجاج القدم وكبر حجم الرأس والمظهر غير الطبيعي وربما انحناء الظهر . قد يعاني الطفل من مشاكل في الانتباه وصعوبة في السمع.13 14 يمكن أن يظهر أحد الأعراض لمرة واحدة فقط، ولكن ظهور مجموعة من الأعراض في نفس الوقت ليس من قبيل الصدفة. ضع كافة الأعراض باعتبارك، واطلب تحويلك إلى أخصائي التمثيل الغذائي عبر طلب ذلك من طبيب عائلتك. في العشرينات والثلاثينيات من العمر، قد يتعرض الشخص البالغ لمشاكل في العظام وصعوبات في الحركة، مثل مشاكل المفاصل والتورم وعدم استقرار المشية والضعف العضلي. وفي النهاية، قد يعتمد المريض على كرسي متحرك، لأنه لم يعد بإمكانه المشي بمفرده.15 قد تكون هناك مشاكل سلوكية أو نفسية، والتي يمكن أن تظهر على شكل نوبات من الارتباك، تكون مصحوبة أحيانًا بالقلق أو الاكتئاب أو الهلوسة.16 سوف يكون العيش المستقل صعبًا بالنسبة لهم، وقد يصبح مرضى نقص ألفا مانوسيداز منعزلين اجتماعيًا.17 18 يصعب توقع الحالة المرضية على المدى البعيد.19
لقد تم اقتراح ثلاثة أنواع فرعية سريرية:37
لكن نظرًا لتنوع الطفرات التي تم توثيقها واتساع نطاق وشدة الأعراض وعدم وجود صلة بين الطفرات المحددة والنوع الفرعي السريري، يُعتبر المرض من الناحية السريرية على أنه سلسلة متصلة من الأعراض التي تتفاوت شدتها.38 39
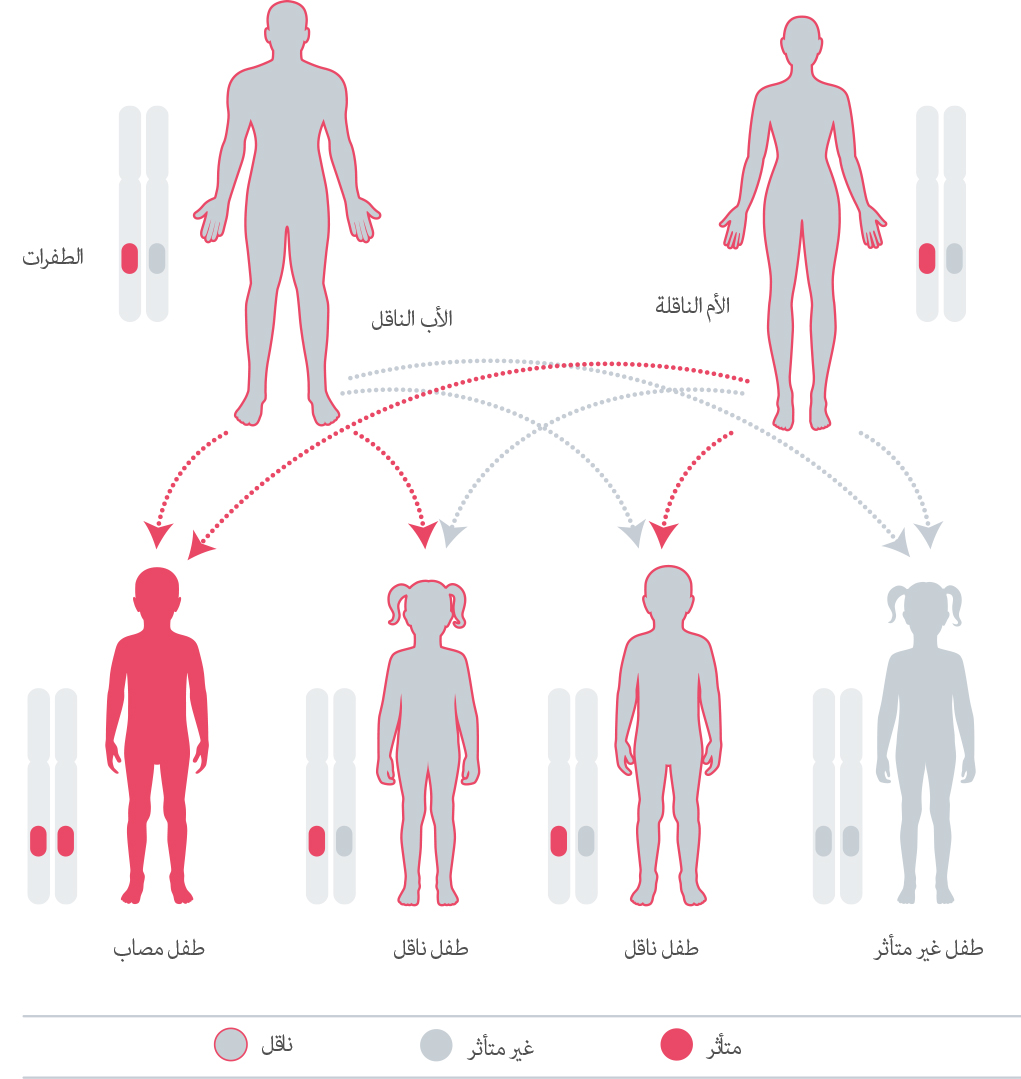
إن مرض نقص ألفا مانوسيداز هو مرض موروث، بمعنى أنه يسري في العائلات.40 حيث نرث الجينات عن والدينا. نحن نرث نسخة من كل جين من كل والد. بعض الجينات التي نرثها تكون جينات”متنحية”، مما يعني أنه يجب توريث كلا نسختي هذا الجين لكي يكون لهما تأثيرًا على تطورنا.41 42 ينتج مرض نقص ألفا مانوسيداز عن جين متنحي.43إ في كل حمل لزوج من الأشخاص يكون كلاهما متغاير الزيجوت لطفرة جين “MAN2B1” المسببة للمرض، تكون هناك فرصة بنسبة 25% لإنجاب طفل مصاب وفرصة بنسبة 50% لإنجاب طفل غير مصاب وحامل للطفرة وفرصة بنسبة 25% لإنجاب طفل غير مصاب وغير حامل للطفرة44 نظرًا لندرة المرض، فإن احتمالية وجود شريك آخر حامل للمرض ضئيلة، ما لم يكون الأفراد من نفس العائلة.45إذا كان الزوجين قد انجبا بالفعل طفلًا يعاني من هذه الحالة، يوصى بالاستشارة الوراثية ، لفهم احتمالات إصابة أبنائهم المستقبليين بالمرض.46
لا نعرف بالضبط مدى انتشار مرض ألفا مانوسيداز. لكن، تقدر عدد من التقارير القادمة من دول مختلفة أن المرض يصيب طفل واحد تقريبًا بين كل مليون مولود في جميع أنحاء العالم.47
يُمكن لطبيبك الاستعانة بعدد من الاختبارات البسيطة للغاية لتشخيص مرض نقص ألفا مانوسيداز، من ضمن تلك الاختبارات:
هناك جمعية دعم حريصة على توفير مصادر المعلومات والتفاهم والنصيحة. كما توجد مجموعة من المنظمات ومجموعات الدعم على بعد نقرات قليلة. منظمات المرضى:
 هي مجموعة دعم دولية لمرض نقص ألفا مانوسيداز. هي مجموعة دعم للعائلات في نيوزيلندا. هي شبكة من مقدمي الرعاية الصحية ومجموعات المرضى في جميع أنحاء أوروبا، وتقدم الدعم للأشخاص المصابين باضطرابات التمثيل الغذائي الموروثة مثل مرض نقص ألفا مانوسيداز.
هي مجموعة دعم دولية لمرض نقص ألفا مانوسيداز. هي مجموعة دعم للعائلات في نيوزيلندا. هي شبكة من مقدمي الرعاية الصحية ومجموعات المرضى في جميع أنحاء أوروبا، وتقدم الدعم للأشخاص المصابين باضطرابات التمثيل الغذائي الموروثة مثل مرض نقص ألفا مانوسيداز.يمكن أن يؤثر تشخيص الإصابة بمرض نقص ألفا مانوسيداز تأثيرًا عاطفيًا كبيرًا على المرضى والقائمين على رعايتهم. يجب توفير إمكانية الحصول على الرعاية الأولية والمتخصصة والخدمات الاجتماعية، كما ينبغي تقييم الدعم الغذائي والنفسي بصفة مستمرة.52 يجب أن تكون الرعاية السريرية استباقية، حيث يخطط الأطباء للحد من عواقب الحالة وأي مضاعفات قد تصيب المريض53 يمكن التفكير في جراحة المفاصل أو جراحة العظام الأخرى.54 للمزيد من المعلومات حول خيارات الإدارة، يرجى مناقشة الأمر مع طبيبك.
– إن الغرض من المعلومات الواردة على الموقع الإلكتروني هو توفير المعرفة بالموضوعات الصحية المتعلقة بمرض نقص ألفا مانوسيداز فحسب. لا ينبغي استخدام هذه المعلومات بدلًا من استشارة طبيبك العام أو أي متخصص أخر في الرعاية الصحية. إذا كانت تساورك الشكوك، يُرجى الاتصال بطبيبك للحصول على النصيحة. تم إنتاج هذا الموقع بواسطة شركة كييزي فامارتشوتشي “CHIESI Farmaceutici “ لقد تم تطوير هذا الموقع الإلكتروني طبقًا للمعايير الصناعية والقانونية لتوفير المعلومات المتعلقة بالموضوعات الصحية لمرض نقص ألفا مانوسيداز لخبراء الرعاية الصحية وعموم الناس. تبذل شركة كييزي فامارتشوتشي “CHIESI Farmaceutici” ما بوسعها من جهود معقولة من أجل تضمين معلومات دقيقة وحديثة. ومع ذلك، إن المعلومات الواردة في هذا الموقع الإلكتروني ليست شاملة.


