Essa doença rara foi descrita pela primeira vez pelo médico sueco Okerman em 1967 e faz parte de uma ampla variedade de distúrbios de depósito lisossômico (DLD), causados por mutações que afetam a enzima lisossômica alfamanosidase, resultando em sua deficiência. A enzima alfamanosidase, que quebra os resíduos de manose dos oligossacarídeos ligados ao N.3
Figura 1a. alfamanosidase cliva os resíduos de manose ligados a alfa dos oligossacarídeos ligados a N.

Figura 1b.
Nas células saudáveis, a alfamanosidase nos lisossomos atua na degradação sequencial de glicoproteínas complexas. Produtos de decomposição menores deixam o lisossomo. Na alfamanosidose, o acúmulo de oligossacarídeos ligados a N ricos em a-manosil leva ao ingurgitamento lisossômico e à interrupção da função celular normal.
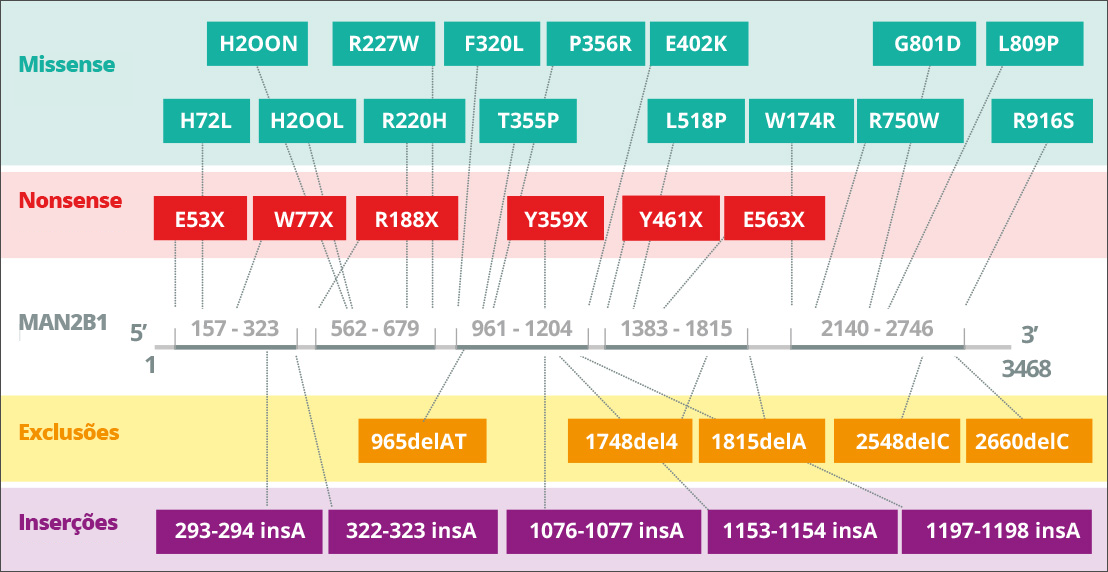
A Alfamanosidose é causada por mutações hereditárias no gene MAN2B1 (LAMAN) que codifica a α-manosidase lisossômica4A. A Alfamanosidose tem herança autossômica recessiva. O gene MAN2B1 é composto por 24 éxons e codifica um polipeptídeo de 1011 aminoácidos que é, de forma pós-translacional, modificado no retículo endoplasmático5A. Durante a maturação e o transporte endossômico do MAN2B1 para os lisossomos, ele é proteoliticamente clivado em três principais polipeptídeos denominados “abc”, “d” e “e” de 70, 42 e 15 kDa, respectivamente6. Mais especificamente, o processamento da subunidade de 70 kDa resulta em um total de cinco polipeptídeos diferentes. O nível de expressão de MAN2B1 parece ser mais alto nos pulmões, rins, pâncreas e nos leucócitos do sangue periférico7A. No SNC, o nível mais alto de expressão parece estar no corpo caloso e na medula espinhal, enquanto níveis consideravelmente mais baixos são observados nas estruturas maiores, que incluem cerebelo, córtex cerebral, lobos frontal e temporal. No entanto, o significado (se houver) de tais variações não está claro no momento7A.
Mutações no gene MAN2B1 levam à perda da atividade lisossômica da alfamanosidase8A. Dependendo da mutação causadora da MAN2B1, proteínas MAN2B1 mutantes foram detectadas em compartimentos subcelulares, como retículo endoplasmático e lisossomos9A. Por exemplo, a proteína pode ser dobrada incorretamente e retida no retículo endoplasmático, ou pode ser dobrada corretamente e transportada para os lisossomos em uma forma inativa10A. Até o momento, 155 variantes de 191 pacientes foram identificadas e parcialmente caracterizadas no nível bioquímico11A.
Se a enzima alfamanosidase estiver prejudicada, ocorre uma redução na degradação das glicoproteínas e um acúmulo progressivo de oligossacarídeos ricos em manose em todos os tecidos, levando à função celular prejudicada e apoptose.13
Alfamanosidose é herdada de forma autossômica recessiva, causada por mutações no gene MAN2B1, localizado no cromossomo 19.
A variabilidade fenotípica é alta, mesmo entre irmãos com genótipos idênticos14. Além de fatores genéticos, fatores ambientais podem influenciar a doença. Por exemplo, a exposição a patógenos pode causar infecções recorrentes e um agravamento dos sintomas da doença.15
As informações contidas neste site destinam-se apenas a fornecer conhecimento dos conceitos de saúde da doença Alfamanosidose. Estas informações não devem ser usadas no lugar de aconselhamento do seu médico ou outro profissional de saúde. Em caso de dúvida, entre em contato com seu médico para obter orientação. Este site foi produzido pela Chiesi Pharmaceuticals. O site foi desenvolvido de acordo com os padrões legais e do setor para fornecer informações aos profissionais de saúde e ao público em geral sobre os tópicos de saúde da doença Alfamanosidose. A Chiesi Pharmaceuticals faz todos os esforços possíveis para incluir informações precisas e atuais. No entanto, as informações fornecidas neste site estão em constante atualização.


