Esta enfermedad rara fue descrita por primera vez por el médico sueco Okerman en 1967, y es uno de los muchos trastornos por almacenamiento lisosomal provocados por mutaciones que afectan a la enzima lisosomal alfa-manosidasa y provocan su déficit. Esta enzima es una exoglicosidasa, que separa los residuos de manosa unidos mediante enlace α de los oligosacáridos unidos con enlace N.3
Figura 1a. La α-manosidasa separa los residuos de manosa unidos mediante enlace alfa de los oligosacáridos unidos con enlace N.

Figura 1b. En las células sanas, la α-manosidasa de los lisosomas interviene en la degradación secuencial de las glucoproteínas complejas. Los productos más pequeños de la descomposición salen del lisosoma. En la α-manosidosis, la acumulación de oligosacáridos con enlace N ricos en α-manosilo tiene como consecuencia la congestión del lisosoma y la alteración del funcionamiento celular normal.
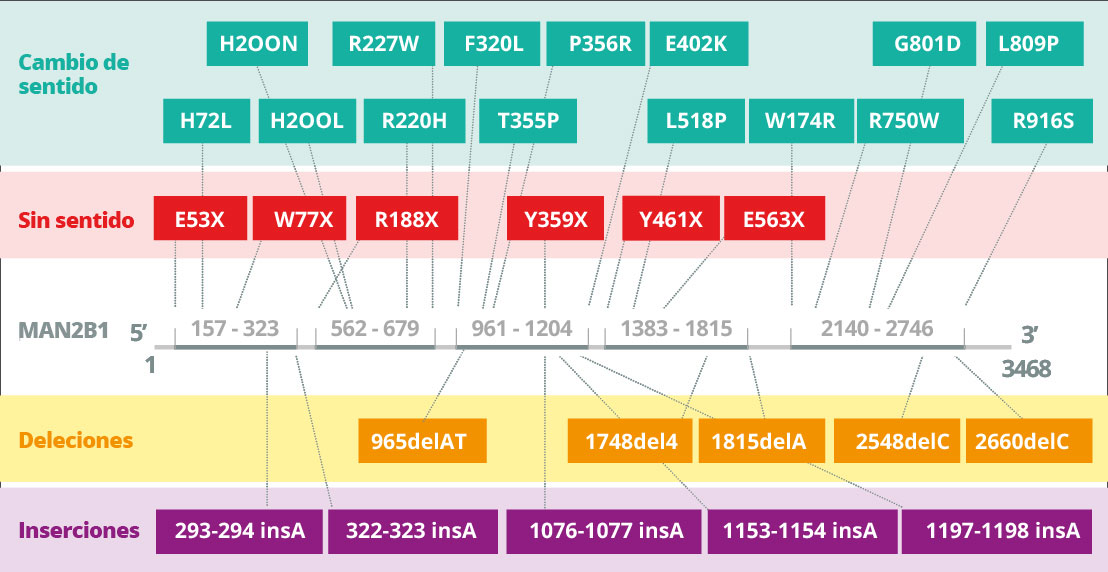
La alfa manosidosis está causada por mutaciones hereditarias del gen MAN2B1 (LAMAN) que codifica la α-manosidasa lisosomal4A. La alfa manosidosis se transmite de forma autosómica recesiva. El gen MAN2B1 está compuesto de 24 exones y codifica un polipéptido de 1011 aminoácidos que experimenta una modificación postraduccional en el retículo endoplasmático5A. Durante la maduración y el transporte endosómico del MAN2B1 a los lisosomas, se separa proteolíticamente en tres polipéptidos principales denominados «abc», «d» y «e», de 70, 42 y 15 kDa, respectivamente6. El procesamiento posterior y más específico de la subunidad de 70 kDa genera en total cinco polipéptidos diferentes. El nivel de la expresión del MAN2B1 parece ser mayor en los pulmones, riñones, páncreas y leucocitos sanguíneos periféricos7A. En el sistema nervioso central, el mayor nivel de expresión parece estar en el cuerpo calloso y la médula espinal, mientras que se observan niveles considerablemente más bajos en las estructuras mayores, que incluyen el cerebelo, la corteza cerebral y los lóbulos frontal y temporal. Sin embargo, la importancia (si hubiere) de dichas variaciones no está clara actualmente7A.
Las mutaciones en el MAN2B1 provocan la pérdida de la actividad de la alfa manosidasa lisosomal8A. En función de la mutación causal del MAN2B1, se han detectado proteínas mutantes del MAN2B1 en orgánulos intracelulares como el retículo endoplasmático y los lisosomas9A. Por ejemplo, la proteína puede plegarse incorrectamente y detenerse en el retículo endoplasmático o puede plegarse correctamente y transportarse a los lisosomas de forma inactiva10A. Hasta la fecha, se han identificado y caracterizado parcialmente 155 variantes de 191 pacientes a nivel bioquímico11A.
Si se bloquea la enzima alfa manosidasa, hay una reducción en la degradación de las glucoproteínas y una acumulación progresiva de oligosacáridos ricos en manosa en todos los tejidos, lo que provoca un deterioro de las funciones celulares y apoptosis.13 La alfa manosidosis se transmite de forma autosómica recesiva, causada por mutaciones del gen MAN2B1, situado en el cromosoma 19. La variabilidad fenotípica es elevada, incluso entre hermanos con genotipos idénticos14. Además de los factores genéticos, la enfermedad puede estar influida por factores ambientales. Por ejemplo, la exposición a patógenos puede causar infecciones recurrentes y un empeoramiento de los síntomas de la enfermedad.15
El objetivo de la información contenida en este sitio web es únicamente dar a conocer temas relacionados con la enfermedad alfa-manosidosis. Esta información no debesustituir el consejo de su médico de cabecera o de otro profesional sanitario. Si tiene dudas, póngase en contacto con su médico. Este sitio web ha sido producido porChiesi Pharmaceuticals. El sitio web ha sido desarrollado de conformidad con los estándares industriales y jurídicos para aportar información a profesionales sanitarios y alpúblico en general sobre temas relacionados con la enfermedad alfa-manosidosis. Chiesi Pharmaceuticals se esfuerza al máximo por incluir información precisa yactualizada. No obstante, la información contenida en este sitio web no es exhaustiva.


