Cette maladie rare a été décrite pour la première fois par le médecin suédois Okerman en 1967, et elle fait partie du grand nombre de maladies lysosomales dues à des mutations qui influent sur l’enzyme lysosomale alpha-mannosidose, ce qui engendre son déficit. Cette enzyme est une exoglycosidase qui sépare les résidus de l’alpha-mannose des oligosaccharides N-liés.3
Figure 1a. L’alpha-mannosidase coupe les résidus de l’alpha-mannose des oligosaccharides N-liés.

Figure 1b. Dans les cellules saines, l’alpha-mannosidase des lysosomes agit par dégradation séquentielle des glycoprotéines complexes. Les plus petits produits de la rupture quittent le lysosome. Dans l’alpha-mannosidose, l'accumulation d’oligosaccharides N-liés riches en alpha-mannose provoque un engorgement lysosomal et l’interruption de la fonction normale de la cellule.
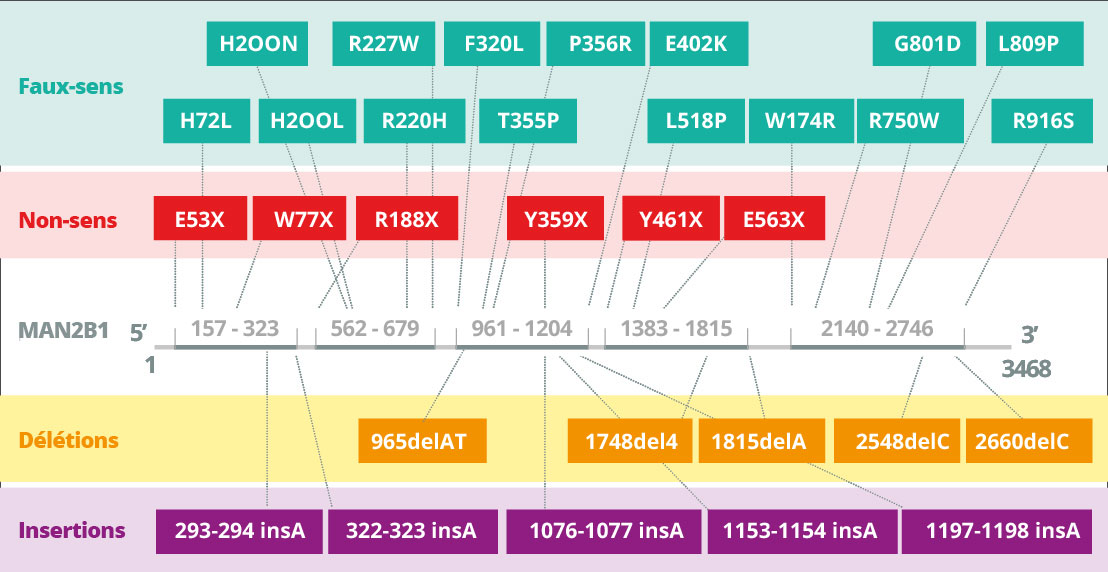
L’alpha-mannosidose est provoquée par des mutations du gène MAN2B1 (LAMAN) qui code l’alpha-mannosidase lysosomale4A. L’alpha-mannosidose est une maladie héréditaire autosomique récessive. Le gène MAN2B1 est composé de 24 exons et codifie un polypeptide de 1011 acides aminés qui sont modifiés après la translation en réticulum endoplasmique5A. Durant la maturation et le transport endosomal du gène MAN2B1 vers les lysosomes, celui-ci est séparé par protéolyse en trois principaux polypeptides appelés « abc », « d » et « e » de 70, 42 et 15 kDa respectivement6. De manière plus spécifique, le traitement de la sous-unité 70 kDa génère au total cinq polypeptides différents. Le niveau de l’expression MAN2B1 est plus élevé dans les poumons, les reins, le pancréas et les leucocytes du sang périphérique7A. Dans le CNS, le niveau le plus élevé de l’expression figure dans le corps calleux et la moelle épinière, tandis qu’un niveau largement plus faible apparaît dans les plus grandes structures, comme le cervelet, le cortex cérébral et les lobes frontaux et temporaux. Toutefois, la signification (si elle existe) de ces variations n’est actuellement pas claire7A.
Les mutations du gène MAN2B1 provoquent la perte d’activité de l’alpha-mannosidase lysosomale8A. Selon la cause de la mutation du gène MAN2B1, les protéines du gène MAN2B1 mutant ont été détectées dans les compartiments sous-cellulaires tels que le réticulum endoplasmique et les lysosomes9A. La protéine peut par exemple être pliée de manière incorrecte et arrêtée dans le réticulum endoplasmique, ou elle peut être pliée correctement et transportée dans les lysosomes sous forme inactive10A. Actuellement, 155 variants génétiques issus de 191 patients ont été identifiés et partiellement caractérisés au niveau biochimique11A.
Si l’enzyme alpha-mannosidase est endommagée, la dégradation des glycoprotéines est réduite et des oligosaccharides riches en mannose s’accumulent progressivement dans tous les tissus, ce qui endommage la fonction cellulaire et conduisant à l’apoptose.13 L’alpha-mannosidose est une maladie héréditaire autosomique récessive provoquée par des mutations du gène MAN2B1, qui se trouve dans le chromosome 19. La variabilité phénotypique est élevée, même chez les frères et sœurs dont le génotype est identique14. Outre les facteurs génétiques, des facteurs environnementaux peuvent influer sur la maladie. L’exposition à des pathogènes peut par exemple provoquer des infections récurrentes et aggraver les symptômes de la maladie.15
Les informations figurant sur ce site web sont uniquement destinées à fournir des connaissances sur les thématiques de santé liées à l’alpha-mannosidose. Ces informations ne doivent pas remplacer les conseils de votre médecin ou d’un autre professionnel de santé. En cas de doute, veuillez contacter votre médecin pour obtenir des conseils. Ce site web a été réalisé par Chiesi Pharmaceuticals. Il a été développé conformément aux normes industrielles et juridiques afin de fournir des informations aux professionnels de santé et au grand public sur des thématiques de santé liées à l’alpha-mannosidose. Chiesi Pharmaceuticals fait tout son possible pour inclure des informations précises et actuelles. Toutefois, les informations fournies sur ce site ne sont pas exhaustives.