Questa malattia rara è stata descritta per la prima volta dal medico svedese Okerman nel 1967 e fa parte dell’ampio spettro di malattie da accumulo lisosomiale, causata da mutazioni che interessano l’enzima lisosomiale alfa-mannosidasi e che ne provocano la carenza. Questo enzima è un’esoglicosidasi che scinde residui di mannosio α-linked di oligosaccaridi N-linked.1
Figura 1a: α-mannosidasi scinde residui di mannosio α-linked di oligosaccaridi N-linked

Figura 1b: nelle cellule sane, l’α-mannosidasi nei lisosomi agisce sulla degradazione sequenziale di glicoproteine complesse. I prodotti di degradazione più piccoli e scissi si separano dal lisosoma. Nell’Alfa-mannosidosi si ha l’accumulo di oligosaccaridi N-linked ricchi di mannosio che porta alla congestione lisosomiale e allo squilibrio della normale funzione cellulare.
L’Alfa-mannosidosi è causata da mutazioni ereditarie del gene MAN2B1 (LAMAN) che codifica per l’α-mannosidasi lisosomiale.2 È una malattia ereditaria, a trasmissione autosomica recessiva. Il gene MAN2B1 è composto da 24 esoni e codifica per un polipeptide di 1011 amminoacidi che viene modificato post-traduzione nel reticolo endoplasmatico.2 Durante la maturazione e il trasporto endosomiale di MAN2B1 ai lisosomi, viene scisso proteoliticamente in tre principali polipeptidi denominati “abc”, “d” ed “e” rispettivamente da 70, 42 e 15 kDa.2 Più nel dettaglio, la modificazione della subunità da 70 kDa determina un totale di cinque diversi polipeptidi. Il livello di espressione di MAN2B1 sembra essere maggiore nei leucociti di polmoni, reni, pancreas e sangue periferico.1 Nel sistema nervoso centrale, il livello di espressione sembra essere massimo nel corpo calloso e nel midollo spinale, mentre livelli considerevolmente più bassi sono osservati nelle strutture più grandi, tra cui cervelletto, corteccia cerebrale, lobi frontali e temporali. Tuttavia, l’eventuale importanza di tali variazioni non è al momento chiara.1
Le mutazioni di MAN2B1 determinano la perdita di attività dell’alfa-mannosidasi lisosomiale.2 A seconda della mutazione di MAN2B1 responsabile, le proteine mutanti MAN2B1 sono state rilevate in compartimenti subcellulari come il reticolo endoplasmatico e i lisosomi.2 Ad esempio, la proteina può essere mal ripiegata e arrestata nel reticolo endoplasmatico oppure può essere ripiegata nel modo corretto e trasportata ai lisosomi in forma inattiva.2 A oggi, sono state identificate e parzialmente caratterizzate a livello biochimico 155 varianti in 191 pazienti.3
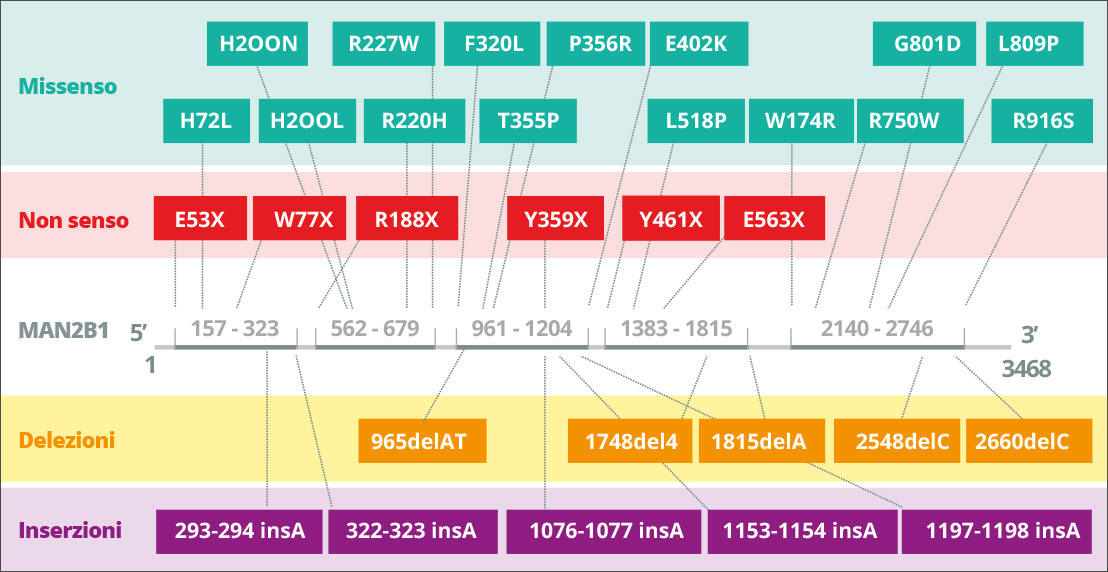
Se l’enzima alfa-mannosidasi è malfunzionante o mancante, si verifica una riduzione nella degradazione delle glicoproteine e un progressivo accumulo di oligosaccaridi ricchi di mannosio in tutti i tessuti che porta alla compromissione della funzione cellulare e all’apoptosi.2 L’Alfa-mannosidosi è una malattia ereditaria a trasmissione autosomica recessiva. È causata da mutazioni nel gene MAN2B1, localizzato nel cromosoma 19. La variabilità fenotipica è alta, anche tra fratelli e sorelle con identici genotipi.1 Oltre ai fattori genetici, anche i fattori ambientali possono influenzare la malattia. Per esempio, l’esposizione ad agenti patogeni può causare infezioni ricorrenti e un peggioramento dei sintomi della malattia.1
Le informazioni che si trovano su questo sito sono volte soltanto a fornire conoscenze riguardanti questioni di salute sulla malattia dell’Alfa-mannosidosi. Queste informazioni non devono essere usate al posto di un consiglio del proprio medico o di un professionista della salute. In caso di dubbi contattare il proprio medico. Questo sito è stato prodotto da Chiesi Farmaceutici. Il sito è stato sviluppato in accordo con gli standard legali e di settore per fornire informazioni ai professionisti della salute e al pubblico in generale sulle tematiche della patologia Alfa-mannosidosi. Chiesi Farmaceutici compie il massimo sforzo per inserire informazioni accurate e aggiornate. Tuttavia, le informazioni qui fornite non sono esaustive.


