L’alfa-mannosidosi è un raro disturbo da accumulo lisosomiale che si manifesta generalmente nella prima infanzia. È una malattia progressiva e fortemente eterogenea, difficile da riconoscere, e di solito si arriva a una diagnosi dopo il consulto con diversi specialisti.1
La maggior parte dei bambini sembra normale alla nascita, ma le manifestazioni cliniche iniziano a partire da un’età molto precoce, seguite da una progressione dei sintomi clinici.2 I pazienti possono presentare un continuum di manifestazioni cliniche.3
La diagnosi di alfa-mannosidosi è resa difficile dalla rarità della malattia e dalla presentazione fortemente eterogenea, per cui spesso viene tralasciata o raggiunta tardi: la malattia è sottodiagnosticata e la prognosi a lungo termine è sfavorevole se non trattata.1
È fondamentale un approccio multidisciplinare nella gestione della malattia, allo scopo di offrire al paziente il trattamento che sia il più possibile personalizzato e mirato a migliorare le sue condizioni o rallentare la progressione della malattia.1
Insieme all’approccio multidisciplinare, la diagnosi precoce rimane il modo migliore per avviare un trattamento tempestivo e ridurre al minimo la progressione dei sintomi.2
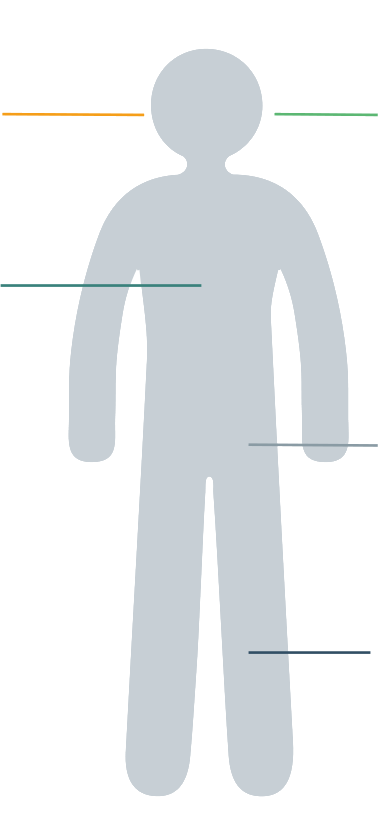
Alcune delle principali caratteristiche cliniche dell’alfa-mannosidosi sono: 3–5





Le informazioni che si trovano su questo sito sono volte soltanto a fornire conoscenze riguardanti questioni di salute sulla malattia dell’Alfa-mannosidosi. Queste informazioni non devono essere usate al posto di un consiglio del proprio medico o di un professionista della salute. In caso di dubbi contattare il proprio medico. Questo sito è stato prodotto da Chiesi Farmaceutici. Il sito è stato sviluppato in accordo con gli standard legali e di settore per fornire informazioni ai professionisti della salute e al pubblico in generale sulle tematiche della patologia Alfa-mannosidosi. Chiesi Farmaceutici compie il massimo sforzo per inserire informazioni accurate e aggiornate. Tuttavia, le informazioni qui fornite non sono esaustive.



