Это редкое заболевание было впервые описано шведским врачом Окерманом в 1967 году и относится к обширной группе лизосомальных болезней накопления, вызванных мутациями в гене, кодирующем лизосомальный фермент альфа-маннозидазу, приводящими к его дефициту. Этот фермент представляет собой экзогликозидазу, которая расщепляет α-связанные остатки маннозы в N-связанных олигосахаридах 3
Рис. 1a. α-маннозидаза отщепляет альфа-связанные остатки маннозы от N-связей олигосахаридов.

Рисунок 1b. В здоровых клетках альфа-маннозидаза в лизосомах отвечает за последовательное расщепление сложных гликопротеинов. Более мелкие продукты расщепления выходят из лизосомы. При α-маннозидозе накопление богатых α-маннозилом N-связанных олигосахаридов приводит к переполнению лизосом и нарушению нормальной функции клеток.
Причиной развития альфа-маннозидоза являются передающиеся по наследству мутации в гене MAN2B1 (LAMAN), кодирующем лизосомальную альфа-маннозидазу 4A. Альфа-маннозидоз наследуется по аутосомно-рецессивному типу. Ген MAN2B1 состоит из 24 экзонов и кодирует 1011-й аминокислотный полипептид, который подвергается посттрансляционной модификации в эндоплазматическом ретикулуме 5A. Во время созревания и эндосомального транспорта белка MAN2B1 в лизосомы он протеолитически расщепляется на три основных полипептида, получивших названия «abc», «d» и «e» и имеющих молекулярную массу 70, 42 и 15 кДа соответственно 6. В ходе дальнейшего процессинга субъединицы массой 70 кДа образуется в общей сложности пять различных полипептидов. Наиболее высокие уровни экспрессии MAN2B1 отмечаются в легких, почках, поджелудочной железе и лейкоцитах периферической крови 7A. В ЦНС самый высокий уровень экспрессии, по-видимому, наблюдается в мозолистом теле и спинном мозге, в то время как значительно более низкие уровни экспрессии имеют место в более крупных структурах, таких как мозжечок, кора головного мозга, лобные и височные доли. Значение (если таковое имеется) различных уровней экспрессии белка в настоящее время не известно 7A.
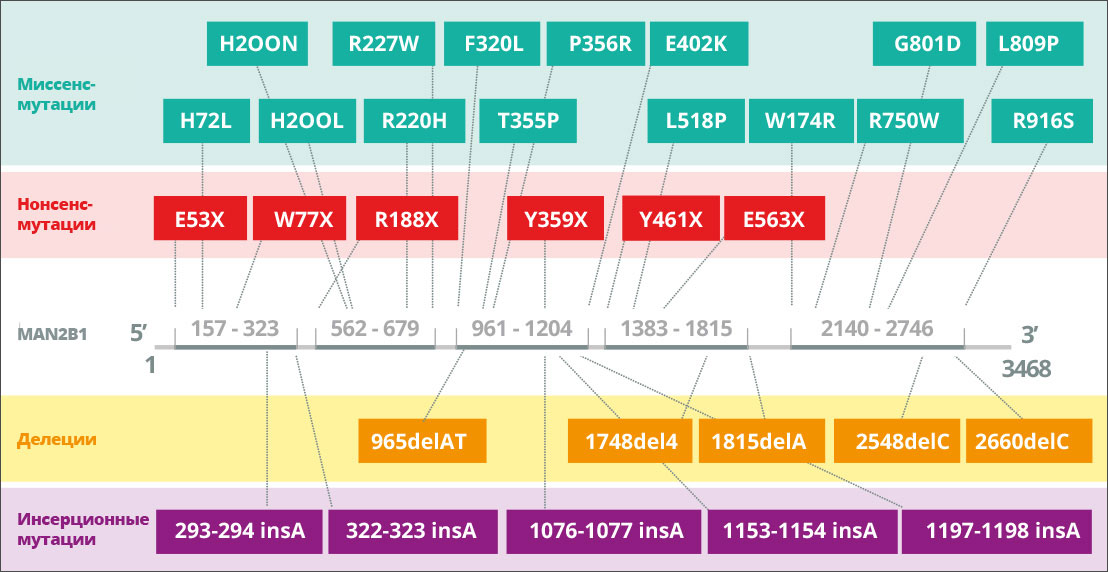
Мутации в гене MAN2B1 приводят к потере активности альфа-маннозидазы в лизосомах 8A. В зависимости от характера мутации в MAN2B1 патологически измененный белок MAN2B1 обнаруживается во внутриклеточных компартментах, таких как эндоплазматический ретикулум и лизосомы 9A. Например, может быть нарушена конформация белка, и он «застревает» в эндоплазматическом ретикулуме, или конформация белка не нарушается, и он транспортируется в неактивном виде в лизосомы 10A. К настоящему времени было выявлено 155 вариантов у 191 пациента, которые были изучены на биохимическом уровне11A.
Если имеет место нарушение функции фермента, это приводит к уменьшению расщепления гликопротеинов и прогрессирующему накоплению богатых маннозой олигосахаридов во всех тканях, что приводит к нарушению функции клеток и апоптозу 13.
Альфа-маннозидоз наследуется по аутосомно-рецессивному типу и вызывается мутациями в гене MAN2B1, расположенном на 19-й хромосоме.
Фенотипическая вариабельность достаточно выражена, даже среди родных братьев и сестер с идентичными генотипами 14. Помимо генетических факторов, на развитие заболевания влияют факторы окружающей среды. Например, контакты с возбудителями инфекционных заболеваний способствуют развитию рецидивирующих инфекций и увеличению выраженности симптомов заболевания 15.
Информация на этом веб-сайте предоставлена в целях ознакомления со свойствами заболевания альфа-маннозидоз. Эта информация не заменяет рекомендации лечащего врача.
Этот веб-сайт создан компанией Кьези Фармацевтичи С.п.А. Веб-сайт разработан в соответствии с отраслевыми и правовыми стандартами для предоставления медицинским работникам и широкой общественности информации о проблемах со здоровьем, связанных с заболеванием альфа-маннозидоз. Компания Кьези Фармацевтичи С.п.А. прилагает все разумные усилия для добавления точной и актуальной информации, однако информация, представленная на этом веб-сайте, не является исчерпывающей.


