Bu nadir hastalık ilk kez 1967’de İsveçli Doktor Okerman tarafından tanımlanmıştır ve alfa-mannosidaz lizozomal enzimini etkileyen mutasyonların neden olduğu ve eksikliğine neden olan çok çeşitli Lizozomal Depolama Bozukluklarından biridir. Bu enzim, N-bağlı oligosakkaritlerin α-bağlı mannoz kalıntılarını parçalayan bir ekzoglikozidazdır.3
Şekil 1a. a-mannosidaz, N-bağlı oligosakaritlerin alfa-bağlı mannoz kalıntılarını parçalar.

Şekil 1b. Sağlıklı hücrelerde, lizozomlardaki α-mannosidaz, kompleks glikoproteinlerin sıralı bozunmasında rol oynar. Daha küçük parçalanma ürünleri lizozomdan ayrılır. α-mannosidozda α-mannosil açısından zengin N-bağlı oligosakkaritlerin birikmesi, lizozomal konjesyona ve normal hücre fonksiyonunun bozulmasına yol açar.
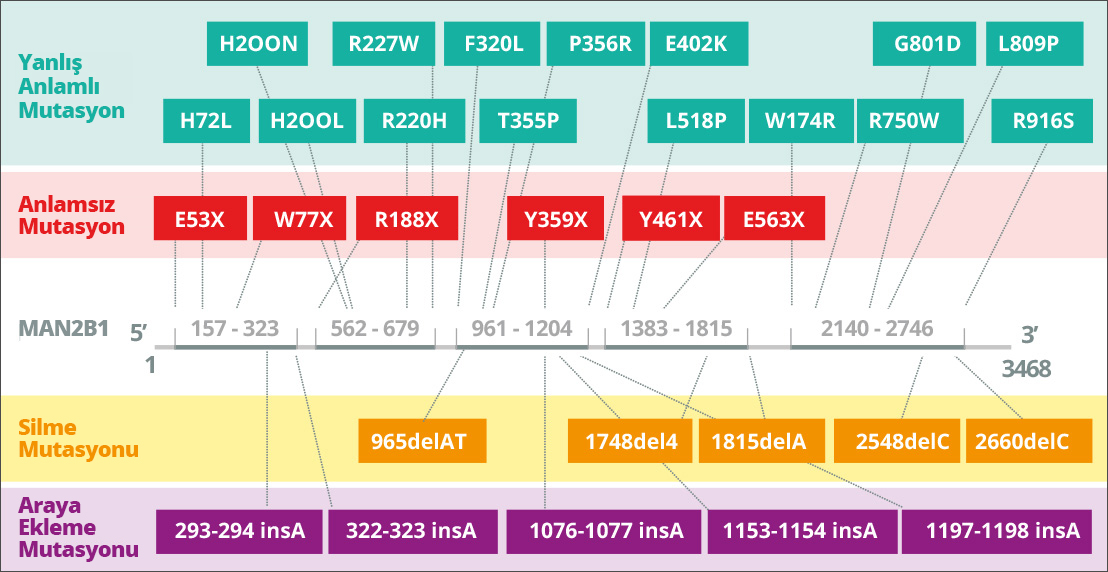
Alfa mannosidaz, lizozomal α-mannosidazı4A kodlayan MAN2B1 (LAMAN) genindeki kalıtsal mutasyonlardan kaynaklanır. Alfa mannosidoz otozomal resesif geçişlidir. MAN2B1 geni 24 ekzondan oluşur ve endoplazmik retikulum5A içinde post-translasyonel olarak modifiye edilen 1011 amino asitlik bir polipeptidi kodlar. MAN2B1’in olgunlaşması ve lizozomlara endozomal taşınması sırasında, proteolitik olarak sırasıyla 70, 42 ve 15 kDa’lık “abc”, “d” ve “e” adlı üç ana polipeptite ayrılır6. Daha spesifik olarak, 70 kDa alt biriminin işlenmesi, toplam beş farklı polipeptit ile sonuçlanır. MAN2B1 ekspresyonunun seviyesi en yüksek akciğer, böbrek, pankreas ve periferik kan lökositlerinde görünüyor7A. CNS’de en yüksek ekspresyon seviyesi korpus kallozum ve omurilikte görülürken, serebellum, serebral korteks, frontal ve temporal lobları içeren daha büyük yapılarda oldukça düşük seviyeler gözlenir. Ancak, bu tür varyasyonların önemi (varsa) şu anda net değil7A.
MAN2B1’deki mutasyonlar, lizozomal alfa mannosidaz aktivitesinin kaybına yol açar8A. Neden olan MAN2B1 mutasyonuna bağlı olarak, endoplazmik retikulum ve lizozomlar9A gibi hücre altı bölmelerde mutant MAN2B1 proteinleri tespit edilmiştir. Örneğin, protein yanlış bir şekilde katlanabilir ve endoplazmik retikulumda tutulabilir veya doğru şekilde katlanabilir ve aktif olmayan bir biçimde10A lizozomlara taşınabilir. Bugüne kadar, 191 hastadan 155 varyant tanımlanmış ve kısmen biyokimyasal düzeyde11A karakterize edilmiştir.
Alfa mannosidaz enzimi bozulursa, glikoproteinlerin bozunmasında bir azalma ve tüm dokularda mannozdan zengin oligosakkaritlerin progresif birikimi olur, bu da hücresel fonksiyon ve apoptoza yol açar.13
Alfa Mannosidoz, 19. kromozomda bulunan MAN2B1 genindeki mutasyonların neden olduğu, otozomal resesif bir şekilde kalıtılır.
Aynı genotipe sahip kardeşler arasında bile fenotipik değişkenlik yüksektir14. Genetik faktörlerin yanı sıra çevresel faktörler de hastalığı etkileyebilir. Örneğin, patojenlere maruz kalmak tekrarlayan enfeksiyonlara ve hastalık semptomlarının kötüleşmesine neden olabilir.15
Bu web sitesindeki bilgiler sadece alfa mannosidoz hastalığı sağlık konuları hakkında bilgi vermek için tasarlanmıştır. Bu bilgiler, aile doktorunuzun veya diğer sağlık uzmanınızın tavsiyeleri yerine kullanılmamalıdır. Eğer şüpheniz varsa, lütfen tavsiye için doktorunuza başvurunuz. Bu internet sitesi, Chiesi Pharmaceuticals tarafından oluşturulmuştur. Bu internet sitesi, sağlık uzmanlarına ve genel halka alfa mannosidoz hastalığı sağlık konuları hakkında bilgi sağlamak için endüstri standartlarına ve yasal standartlara uygun olarak geliştirilmiştir. Chiesi Pharmaceuticals, doğru ve güncel bilgileri dahil etmek için her makul çabayı göstermektedir. Ancak, bu sitende verilen bilgiler ayrıntılı değildir.


